HomeEnfermedades intestinalesAnálisis a nivel de cepa de Escherichia coli basado en la metagenómica a partir de una serie temporal de muestras de microbioma de un paciente con enfermedad de Crohn
Análisis a nivel de cepa de Escherichia coli basado en la metagenómica a partir de una serie temporal de muestras de microbioma de un paciente con enfermedad de Crohn
Fang Xin , Monje Jonathan M. , Nurk Sergey , Akseshina Margarita , Zhu Qiyun , Gemmell Christopher , Gianetto-Hill Connor , Leung Nelly , Szubin Richard , Sanders Jon , Beck Paul L. , Li Weizhong , Sandborn William J. , Gray-Owen Scott D. , Caballero Rob , Allen-Vercoe Emma , Palsson Bernhard O. , Smarr Larry
La disbiosis del microbioma intestinal, incluida la elevada abundancia de supuestos desencadenantes bacterianos principales, como E. coli, en pacientes con enfermedad inflamatoria intestinal (EII), es de gran interés. Hasta la fecha, la mayoría de los estudios de E. coli en pacientes con EII se centran en aislamientos clínicos, pasando por alto su abundancia relativa y su recambio a lo largo del tiempo. Los estudios basados en la metagenómica, por otro lado, se centran menos en las investigaciones a nivel de cepa. Aquí, utilizando herramientas bioinformáticas recientemente desarrolladas, analizamos la abundancia y las propiedades de cepas específicas de E. coli en un paciente con enfermedad de Crohn (EC) de forma longitudinal, teniendo en cuenta también la composición de toda la comunidad a lo largo del tiempo. En este informe, llevamos a cabo un estudio piloto sobre el análisis metagenómico a nivel de cepa de una serie temporal de cepas de E. coli en un paciente con EC del lado izquierdo, que exhibió niveles sostenidos de E. coli superiores a 100 veces los controles sanos. Nosotros: (1) mapeamos la composición del microbioma intestinal a lo largo del tiempo, en particular la presencia de cepas de E. coli, y descubrimos que la abundancia y el dominio de cepas específicas de E. coli en la comunidad variaban con el tiempo; (2) realizó ensamblajes de novo a nivel de cepa de siete cepas dominantes de E. coli, e ilustró la disparidad entre estas cepas tanto en el origen filogenético como en el contenido genómico; (3) observó que la cepa ST1 (recuperada durante el pico de inflamación) es muy similar a las cepas patógenas conocidas de AIEC NC101 y LF82 tanto en factores de virulencia como en funciones metabólicas, mientras que otras cepas (ST2-ST7) que se recolectaron durante estados más estables mostraron características diversas; (4) aislaron, secuenciaron y caracterizaron experimentalmente ST1 y confirmaron la precisión del ensamblaje de novo; y (5) evaluó la capacidad de crecimiento de ST1 con un modelo metabólico a escala genómica recientemente reconstruido de la cepa, y mostró su potencial para utilizar sustratos que se encuentran abundantemente en el intestino humano para superar a otros microbios. En conclusión, es probable que el estado de inflamación (evaluado por la proteína C reactiva en sangre y la calprotectina en heces) se correlacione con la abundancia de un subgrupo de cepas de E. coli con rasgos específicos. Por lo tanto, el análisis de series temporales a nivel de cepa de las cepas dominantes de E. coli en un paciente con EC es muy informativo y motiva un estudio de una cohorte más grande de pacientes con EII.
1. Introducción
La disbiosis del microbioma intestinal en pacientes con enfermedad inflamatoria intestinal (EII) se asocia con una reducción de la diversidad bacteriana, un aumento de la abundancia relativa de Proteobacteria (Mukhopadhya et al., 2012) y una disminución de Firmicutes (Matsuoka y Kanai, 2015). Específicamente, E. coli se considera una de las posibles causas de formación y progresión de la EII (Rhodes, 2007; Sasaki et al., 2007). Un patotipo específico, la E. coli adherente-invasiva (AIEC), que es capaz de adherirse a las células epiteliales intestinales y sobrevivir y replicarse dentro de los macrófagos, ha sido implicado en la inflamación intestinal (Darfeuille-Michaud et al., 1998; Palmela et al., 2017). Los miembros de este patotipo, así como otros aislados de E. coli asociados a la EII, pertenecen principalmente al filogrupo B2 (Petersen et al., 2009), portando un conjunto diverso de factores de virulencia y mostrando fenotipos metabólicos distintos (Martínez-Medina y García-Gil, 2014; Fang et al., 2018). Sin embargo, no se ha identificado un determinante genético único para este grupo (O’Brien et al., 2016).
Los estudios previos sobre E. coli en la EII se centraron principalmente en aislamientos clínicos extraídos de biopsias intestinales y muestras fecales, que luego se cultivan y caracterizan experimentalmente (Eaves-Pyles et al., 2008; Vejborg et al., 2011; Desilets et al., 2016; O’Brien et al., 2016). Sin embargo, la mayoría de estos estudios no tuvieron en cuenta otros factores, como la composición del microbioma intestinal y la dinámica de la comunidad. Recientemente, con la caída de los costes de secuenciación, los datos metagenómicos se han convertido en una fuente popular de información con la que investigar la composición (Pascal et al., 2017), la función (Morgan et al., 2012; Ni et al., 2017) y dinámica (Halfvarson et al., 2017; Schirmer et al., 2018) del microbioma de la EII. Sin embargo, estos estudios carecen de una caracterización detallada de la comunidad de E. coli. Por lo general, sólo examinan la abundancia relativa de E. coli, pasando por alto la composición a nivel de cepa y los rasgos específicos de la cepa de la comunidad de E. coli, sin embargo, estudios anteriores ya han demostrado la diversidad genética y la variación temporal en la población de E. coli (Caugant et al., 1981).
Afortunadamente, el análisis a nivel de cepa de los datos metagenómicos ha sido posible gracias a herramientas bioinformáticas desarrolladas recientemente, como MIDAS, que caracteriza la variación a nivel de cepa (Nayfach et al., 2016), DESMAN, que permite la extracción de novo de cepas (Quince et al., 2017), entre otras herramientas de genómica de poblaciones a nivel de cepa (Luo et al., 2015; Fischer et al., 2017; Truong et al., 2017). Además, las herramientas desarrolladas para el análisis a nivel de genoma, como los modelos metabólicos a escala genómica (GEM), permiten un análisis exhaustivo a nivel de cepa. Los GEM son reconstrucciones de la red metabólica de cepas que posteriormente se convierten en modelos matemáticos computables, lo que permite mapear entre la base genética y las funciones metabólicas fenotípicas (McCloskey et al., 2013). Debido al contenido genómico versátil de E. coli (Rasko et al., 2008), el análisis GEM a nivel de cepa ha demostrado ser esencial e informativo (Monk et al., 2013).
Aquí, realizamos un estudio piloto en un paciente con EII, específicamente un paciente con enfermedad de Crohn (EC) del lado izquierdo, y realizamos un análisis a nivel de cepa basado en la metagenómica de la comunidad de E. coli de series temporales de pacientes. No solo examinamos la composición del microbioma intestinal, la abundancia relativa de E. coli y la dinámica de la comunidad, sino que también realizamos un análisis a nivel de cepa para identificar, ensamblar y caracterizar las cepas dominantes de E. coli en diferentes momentos, seguido de la validación experimental.
2. Resultados y discusión
2.1. Se recogieron muestras de heces de series temporales y se secuenciaron durante 3 años
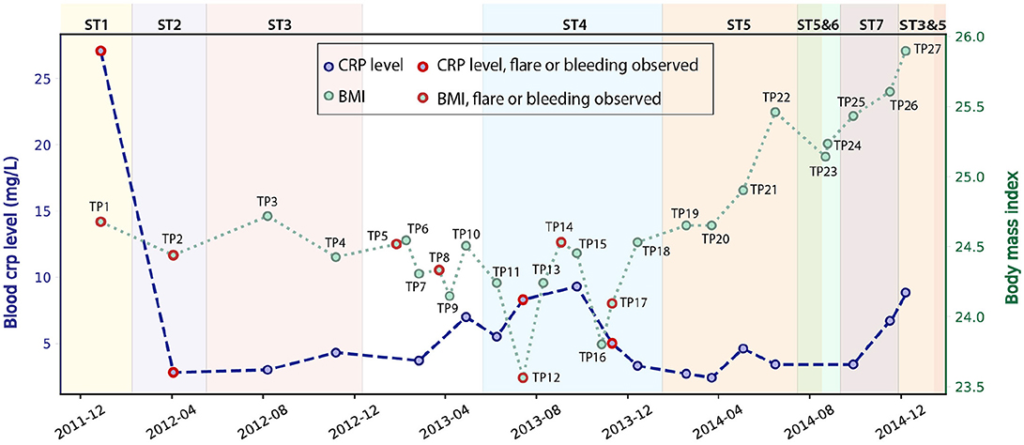
Se estudiaron 27 muestras de heces de series temporales (denominadas TP1-TP27 como se muestra en la Figura 1) recogidas de un paciente masculino con EC de 69 años, que fue diagnosticado de EC colónica a los 63 años con inflamación confinada a su colon sigmoide. Estas muestras se recolectaron durante un período de 3 años entre 2011 y 2014 (Tabla S1), cubriendo tanto estados estables como inflamados (Wu et al., 2013). Generamos datos metagenómicos para cada muestra recolectada y registramos metadatos detallados que incluyen el índice de masa corporal (IMC), el nivel de proteína C reactiva (PCR) en sangre, el nivel de calprotectina fecal y otras mediciones de biomarcadores durante este período (Tabla S1). Durante los 3 años, este paciente tomó Ciprofloxacino, Metronidazol y Prednisona diariamente en febrero de 2012, y también usó Lialda y Uceris de junio a noviembre de 2013. El IMC se registró en todas las muestras y osciló entre 23,6 y 25,9 (Figura 1). Se midió el nivel de PCR de alta sensibilidad (PCR-us), que es indicativo del nivel de inflamación, en 18/27 muestras, y fluctuó entre 2,4 y 27,1 mg/L (Figura 1). La calprotectina fecal mostró una tendencia similar a la de la PCR-hs en sangre, con variación significativa (Figura S2). En particular, los niveles de inflamación en sangre y heces fueron los más altos cuando se recogió la primera muestra, con una PCR-us que alcanzó un máximo de 27,1 mg/L, mientras que el rango normal de PCR-us para los controles sanos es de ≤ de 1 mg/L (Mosli et al., 2015), y con la calprotectina alcanzando un máximo de 2500, más de 50 veces el límite superior para los controles sanos. Por lo tanto, nuestro objetivo fue explorar la relación entre el estado de inflamación y los microbios intestinales, especialmente con la comunidad de E. coli en el microbioma intestinal.
Figura 1
Figura 1. El nivel de PCR-us en sangre y el IMC del paciente fluctuaron durante los 3 años de este estudio (PCR-us solo disponible para 18 muestras). Las muestras recolectadas durante el sangrado o el brote están etiquetadas en rojo. La cepa dominante de E. coli varió para diferentes puntos de tiempo (discutidos en el siguiente párrafo) y están etiquetadas por diferentes colores de fondo.
2.2. La composición del microbioma intestinal y de la comunidad de E. coli ha cambiado a lo largo del tiempo
El análisis de la composición y riqueza del microbioma intestinal indica que la comunidad microbiana intestinal de este paciente fue disbiótica y altamente dinámica durante los 3 años de este estudio. Se realizó la asignación taxonómica de las muestras metagenómicas utilizando MetaPhlan2 (Truong et al., 2017), y se calculó la diversidad alfa y beta de las 27 muestras. En comparación con el microbioma intestinal de los controles sanos, que en su mayoría están dominados por Firmicutes (49-76%) y Bacteroidetes (16-23%) (Matsuoka y Kanai, 2015) con un componente menor de Proteobacteria (mediana = 1%) (Bradley y Pollard, 2017), este paciente tenía un nivel elevado de Proteobacteria que oscilaba entre el 1,09 y el 55,3 %, y un nivel reducido de Firmicutes entre el 22,3 y el 49,1 %. También encontramos fagos de enterobacterias K1E (accesión: NC_007637.1) y K1-5 (accesión: NC_008152.1) en TP1, que no se muestran en los resultados de MetaPhlan2 en (Figura 2) (ver Material suplementario para un análisis detallado). También realizamos un análisis de coordenadas principales (PCoA) sobre la diversidad beta calculada (ver Figura S3) para evaluar la disimilitud entre las muestras.
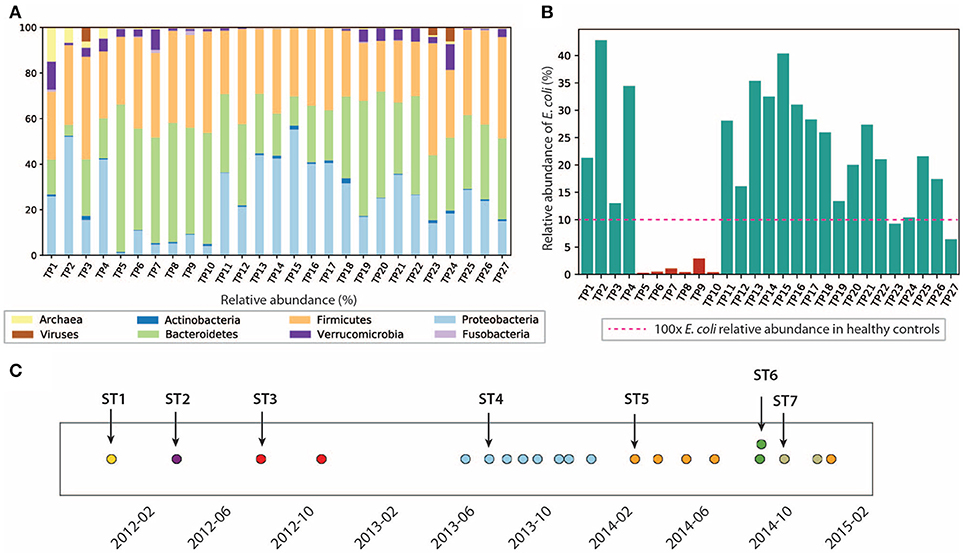
Figura 2
Figura 2. La composición del microbioma intestinal y de la comunidad de E. coli es dinámica. (A) Abundancia relativa de microbios a nivel de filos. (B) Abundancia relativa de E. coli en este paciente. La abundancia relativa de E. coli es de <0,1% en la cohorte sana. (C) Cepas dominantes de la comunidad de E. coli identificadas en 21/27 muestras. Los colores representan diferentes cepas dominantes. Las flechas resaltan las muestras que seleccionamos para su posterior análisis sobre las cepas dominantes.
En particular, caracterizamos la comunidad de E. coli en el microbioma intestinal, ya que E. coli se considera uno de los principales desencadenantes bacterianos de la EII (Rhodes, 2007). La abundancia relativa de E. coli en este paciente oscila entre el 0,1 y el 42,6%, lo que fue anormalmente alto (hasta 400 veces) en comparación con el de los controles sanos (≤0,1% en la cohorte sana Human Microbiome Project Consortium, 2012, pero consistente con la elevada abundancia de E. coli observada en estudios previos de EII (Matsuoka y Kanai, 2015). Durante los 3 años de estudio, el nivel de E. coli se mantuvo relativamente alto, excepto durante los primeros 4 meses de 2013, durante los cuales se recolectaron TP5-TP10 (resaltados en rojo en la Figura 2B). El nivel de inflamación durante este período en particular no mostró diferencias significativas en comparación con otros puntos temporales. Curiosamente, la abundancia relativa de E. coli no se correlacionó necesariamente con el nivel de inflamación en todas las muestras. Por ejemplo, TP2 tiene la mayor abundancia relativa de E. coli de 42,6%, pero solo tiene un nivel de hs-CRP de 2,8 mg/L (1/10 del nivel de hs-CRP para TP1). Dado que E. coli es una especie muy versátil con un pangenoma abierto (Snipen et al., 2009), es posible que solo un subconjunto de cepas de E. coli con ciertas características patógenas contribuyan a la progresión de la enfermedad en la EII. Por lo tanto, investigamos más a fondo la composición a nivel de cepa para la comunidad de E. coli en las 21 muestras que tienen una abundancia relativa de E. coli del ≥5% (resaltado en verde en la Figura 2B). Seis muestras (TP5-TP10) fueron excluidas de estudios adicionales de E. coli debido a su escasez de E. coli.
El análisis de variantes de un solo nucleótido (SNV) en las 21 muestras seleccionadas sugiere que la comunidad de E. coli estaba dominada por una sola cepa en la mayoría de las muestras, y la cepa dominante cambió con el tiempo. Las frecuencias de SNV para especies de E. coli fueron detectadas por MIDAS (Nayfach et al., 2016). La mayoría de las frecuencias de SNV están cerca de 0 o 1 (Figura S4), lo que implica que una sola cepa dominaba típicamente la comunidad de E. coli en un momento dado. Este resultado es consistente con el hallazgo de un estudio anterior de que una sola cepa domina la mayoría de las especies en el microbioma intestinal (Truong et al., 2017).
Las posiciones de los SNV detectados en múltiples muestras también sugieren que la cepa dominante de E. coli cambió con el tiempo (ver sección 5) (Figura 2C), posiblemente debido a alteraciones en la dieta, la estructura ecológica del microbioma y el medio ambiente (incluidos los componentes del sistema inmunitario humano). En las 21 muestras con mayor abundancia relativa de E. coli, identificamos un total de siete cepas dominantes (algunas de ellas abundantes en varios puntos temporales). Para caracterizar mejor las cepas dominantes y comprender su asociación con la inflamación, nos centramos en las muestras resaltadas en la Figura 2C que contienen las siete cepas dominantes.
2.3. Cepas dominantes de E. coli ensambladas y caracterizadas computacionalmente
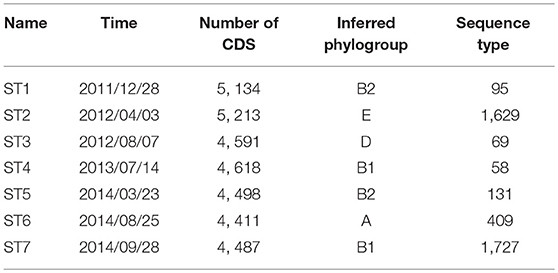
Intentamos recuperar secuencias genómicas de las siete cepas dominantes de E. coli a partir de las muestras seleccionadas. Los ensamblajes preliminares de las cepas dominantes (denominadas ST1-ST7) se obtuvieron mediante ensamblaje metagenómico de novo y agrupamiento de muestras individuales (ver sección 5), seguido de una anotación funcional utilizando Prokka (Seemann, 2014). El número de genes codificantes de proteínas en las anotaciones resultantes oscila entre 4.411 y 5.213 (Tabla 1). Además, realizamos un análisis filogenético utilizando PhyloPhlan (Segata et al., 2013) para inferir el filogrupo de cada ensamblaje. Aunque estudios previos han demostrado que las cepas de los filogrupos B2 y D se encuentran con mayor frecuencia en pacientes con EII (Kotlowski et al., 2007), las siete cepas dominantes en este paciente tienen diversos orígenes filogenéticos y se predice que abarcan los filogrupos B2, E, D, B1 y A. En particular, es probable que ST1 y ST5 pertenezcan al filogrupo B2, que contiene la mayoría de las cepas de AIEC. Además, también hemos asignado los tipos de secuencia de las cepas dominantes utilizando los ensamblajes de novo y BacWGSTdb (Ruan y Feng, 2016). Se ha informado que las cepas dominantes tienen diferentes tipos de secuencia (Tabla 1). Específicamente, el tipo de secuencia 95, 69 y 131 son predominantes en cepas de E. coli patógenas extraintestinales (Doumith et al., 2015).
Tabla 1
Tabla 1. Características de las siete cepas dominantes recuperadas de muestras metagenómicas.
Para explorar más a fondo la diversidad de las cepas seleccionadas, construimos un pangenoma para los siete ensamblajes y encontramos una variación significativa entre las cepas. Construimos el pangenoma con Roary (Page et al., 2015) utilizando un umbral del 80% para la similitud génica (ver sección 5). Identificamos un total de 8.459 ortólogos, de los cuales solo el 37,7% son genes centrales compartidos entre todas las cepas. Entre el resto de los genes accesorios, el 39,9% son exclusivos de una sola cepa (Figura S5), lo que pone de manifiesto la diversidad de las siete cepas. Para explorar más a fondo la variación entre cepas, investigamos las características genómicas y las funciones metabólicas de las cepas dominantes.
2.4. El análisis de las cepas recuperadas revela una diversidad de factores de virulencia
Examinamos la distribución de los factores de virulencia en los siete conjuntos. A modo de comparación, incluimos dos cepas de AIEC bien estudiadas, NC101 (Allen-Vercoe y Jobin, 2014; Ellermann et al., 2015), asociada a la inducción de cáncer de colon (Arthur et al., 2012), y LF82, una cepa de E. coli asociada a pacientes con EC ileal derecha (Darfeuille-Michaud, 2002; Darfeuille-Michaud et al., 2004; Miquel et al., 2010). Además, incluimos la cepa comensal K-12 MG1655, ampliamente estudiada, como cepa de referencia bien definida. En primer lugar, mapeamos los siete ensamblajes del genoma y las tres cepas de referencia en una base de datos de factores de virulencia seleccionada VFDB (Chen et al., 2005) utilizando BLAST (Boratyn et al., 2012) con un umbral del 80% de similitud de secuencia. Este procedimiento identificó un total de 164 factores de virulencia entre las diez cepas (Figura S6). Muchos de estos factores de virulencia están implicados en funciones que previamente están implicadas en la fisiopatología de la EII, como la adquisición de hierro (Dogan et al., 2014), la adhesión (Barnich et al., 2007), los sistemas de secreción (Nash et al., 2010) y la síntesis de cápsulas (Martínez-Medina et al., 2009). Observamos que las cepas del filogrupo B2 (NC101, LF82, ST1, ST5) generalmente tienen más factores de virulencia en comparación con las otras cepas, y tienen más factores de virulencia en común.
2.5. Presencia/ausencia de 57 factores de virulencia conocidos asociados a la EII en las cepas recuperadas
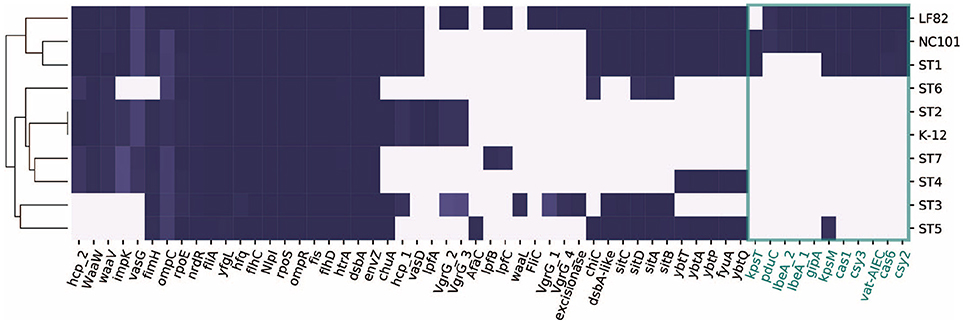
A continuación, nos centramos en 57 genes que se han asociado con la patogenicidad en pacientes con EII de estudios previos (Tabla S2). Recogimos los genes y sus secuencias de la literatura, y los comparamos con las diez cepas utilizando BLAST (Boratyn et al., 2012). Curiosamente, solo ST1 se agrupó con las cepas representativas de AIEC LF82 y NC101, mientras que ST5 no compartió tantos genes con las cepas patógenas seleccionadas (Figura 3). Esto podría explicar por qué el ST1 se correlacionó con un alto nivel de inflamación, mientras que la PCR-us era de solo 2,4 mg/L cuando se recogió el ST5. Encontramos un conjunto de genes que son únicos o más prevalentes en ST1, LF82 y NC101 que los diferencian de otras cepas (resaltados en la Figura 3). Además de los factores de virulencia asociados a la EII, también encontramos que, al igual que NC101, ST1 también alberga la isla genotóxica de la policétido sintasa (pks) que se demostró que induce cáncer colorrectal (Arthur et al., 2012).
Figura 3
Figura 3. Distribución de 57 genes implicados en la patogénesis de AIEC en diez cepas. Los genes exclusivos de ST1, NC101 y LF82 están involucrados en varias funciones, incluida la síntesis de cápsulas (kpsT, Martinez-Medina et al., 2009), la proteasa de mucinas (vat-AIEC,Gibold et al., 2016), los genes asociados a CRISPR (cys3, cas6, cys2 y cas1, Zhang et al., 2015), la invasión (ibeA y su variante, Cieza et al., 2015), las FV codificadas por fagos (gipA, Vazeille et al., 2016), y la utilización de propanodiol (pduC, Dogan et al., 2014).
2.6. Las redes metabólicas diferencian las cepas ST1 y AIEC de otras cepas dominantes recolectadas durante períodos de baja inflamación
Además de los factores de virulencia, también delineamos las diferencias en el metabolismo entre cepas. Construimos redes metabólicas preliminares para siete ensamblajes y las tres cepas de referencia basándonos en los modelos metabólicos a escala genómica (GEM) de múltiples cepas publicados anteriormente (Monk et al., 2013) (ver sección 5). Para las diez redes metabólicas reconstruidas, hay 3.077 reacciones metabólicas en total, entre las cuales 302 son reacciones accesorias que faltan en al menos una cepa, y 2.775 reacciones centrales que están presentes en todas las cepas.
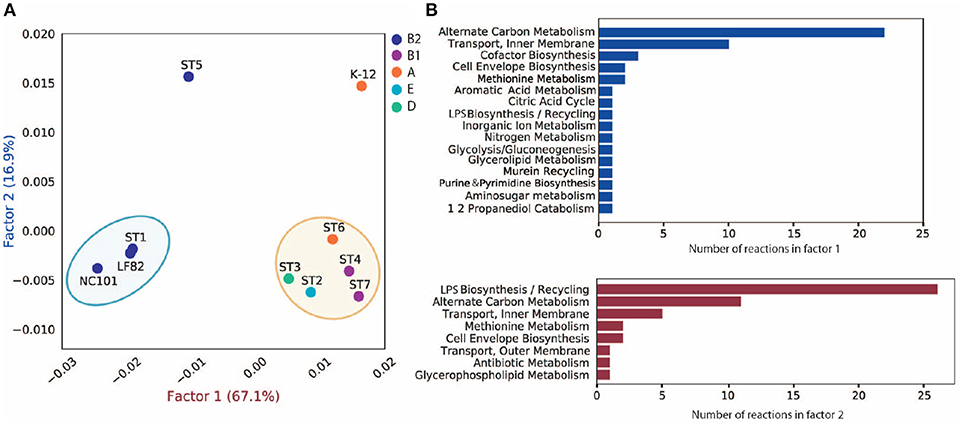
Para investigar la discrepancia en las funciones metabólicas entre estas cepas, creamos un pan-reactoma para estas diez cepas (ver sección 5). A continuación, realizamos un análisis de correspondencias múltiples (MCA) en la matriz pan-reactoma formada por llamadas de ausencia/presencia para estas reacciones, que ha demostrado clasificar eficazmente los reactomas (Monk et al., 2014). A continuación, nos centramos en el factor 1 y el factor 2 (Figura 4A), ya que explicaban un total de 84% de varianza (67,1 y 16,9%, respectivamente).
Figura 4
Figura 4. Análisis MCA del pan-reactoma para diez cepas. (A) Visualización de los resultados del factor 1 y del factor 2 de la ACM. (B) Distribución funcional de reacciones importantes en el factor 1 y el factor 2.
El gráfico del factor 1 vs. el factor 2 (Figura 4A) muestra que TP1 es muy similar a NC101 y LF82 en términos de funciones metabólicas, mientras que las cepas aisladas de otros puntos temporales mostraron características diversas (Figura 4A). Observamos que el factor 1 separó las cepas B2 de las cepas no B2, mientras que el factor 2 separó a TP5 y K12 de las otras cepas. Investigamos más a fondo las 50 reacciones que tienen la mayor contribución a los factores 1 y 2 (Tabla S3), y trazamos su distribución funcional (Figura 4B). Muchas de las principales reacciones que contribuyen al factor 1 están involucradas en el metabolismo alternativo del carbono, la biosíntesis de cofactores y las reacciones de transporte. Un análisis posterior mostró que las cepas B2 y no B2 tienen distintas reacciones involucradas en la utilización de carbono y el transporte de metabolitos (Figura S7A), lo que sugiere que las cepas B2 y las cepas no B2 pueden estar adaptadas a diferentes microambientes y sustratos de nutrientes.
En cuanto a las principales reacciones que contribuyen al factor 2, aunque algunas también están implicadas en el metabolismo del carbono y en las reacciones de transporte, más de la mitad de las reacciones están implicadas en la biosíntesis y el reciclaje de lipopolisacáridos (LPS). Un análisis adicional mostró que TP5 y K12 tienen un conjunto único de reacciones involucradas en la síntesis de LPS en comparación con las otras ocho cepas (Figura S7B). Estudios previos demostraron que la endotoxicidad de los LPS producidos por la microbiota intestinal desempeña un papel vital en el desarrollo de la colitis intestinal (Gronbach et al., 2014). Por lo tanto, la diferencia que observamos en la biosíntesis de LPS puede correlacionarse con el estado de inflamación del huésped y debe estudiarse experimentalmente en el futuro.
El análisis de MCA del pan-reactoma mostró similitud en las funciones metabólicas entre las cepas ST1 y AIEC LF82 y NC101, lo que sugiere que las cepas de E. coli asociadas con inflamación intestinal en pacientes con EII pueden compartir ciertas capacidades metabólicas. Sin embargo, debido a que solo obtuvimos ensamblajes de novo que están incompletos, no pudimos construir GEM precisos para evaluar más a fondo sus capacidades de crecimiento. Para verificar nuestros resultados y permitir una simulación precisa de GEM de la cepa ST1 más interesante, se procedió a su aislamiento, secuenciación y caracterización experimental.
2.7. Aislamiento y caracterización de ST1
Dado que ST1 estaba presente en gran abundancia durante el pico de inflamación y mostraba la mayor semejanza con las cepas AIEC conocidas, se procedió a aislar ST1 de la muestra de heces y caracterizarla experimentalmente. Su identidad se confirmó con el análisis de SNV (ver sección 5). Esta cepa, a la que llamamos CG1MAC, fue secuenciada y ensamblada para dar un genoma de 5.169.659 pb con 4.916 regiones codificantes, de las cuales 4.905 genes estaban presentes en el ensamblaje ST1. La precisión del ensamblaje ST1, en comparación con CG1MAC, es del 95,5%. El análisis genómico adicional mostró que CG1MAC está estrechamente relacionado con 3_2_53FAA (que comparte 4837/4916 ORF), una cepa de E. coli previamente aislada del colon descendente izquierdo inflamado de un paciente masculino con EC de 52 años, y forma parte de la colección del genoma de referencia de HMP con el número de identificación de cepa HM-38 (Human Microbiome Jumpstart Reference Strains Consortium et al., 2010) (ver Material Complementario). Notamos la similitud en el sexo, la edad y el sitio de inflamación del colon con nuestro paciente. Además, el Laboratorio Nacional de Microbiología de Canadá determinó experimentalmente que el serotipo de CG1MAC era O2:H7. Los análisis filogenéticos sugieren que CG1MAC está evolutivamente estrechamente relacionado con AIEC y cepas uropatógenas (UPEC) en el filogrupo B2 (Figura S8).
Para examinar si CG1MAC presentaba características AIEC, realizamos ensayos de adhesión e invasión. Los resultados experimentales mostraron que CG1MAC es capaz de adherirse bien a la línea celular epitelial intestinal Caco-2, pero no invade los macrófagos THP-1, a diferencia de la cepa representativa de AIEC LF82. CG1MAC fue engullido a un nivel bajo y mostró una supervivencia intracelular deficiente (ver Material suplementario).
2.8. Se prevé que la capacidad de crecimiento de CG1MAC sea similar a la de las cepas AIEC
Construimos un borrador de modelo a escala genómica (GEM) para CG1MAC basado en su secuencia genómica y modelos de E. coli publicados anteriormente (Monk et al., 2013) (ver sección 5). El GEM de la cepa CG1MAC contiene 1.581 genes, 2.913 reacciones metabólicas y 2.115 metabolitos. A continuación, predijimos la capacidad de crecimiento de CG1MAC, junto con tres modelos de referencia de borrador K-12, LF82 y NC101 que se reconstruyeron siguiendo el mismo procedimiento.
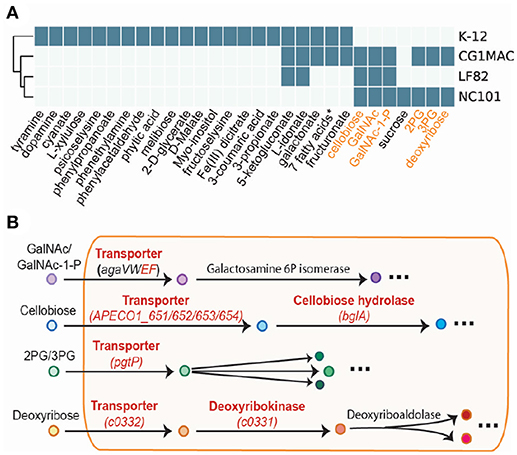
Los resultados de la simulación de crecimiento en varias fuentes de nutrientes indican que CG1MAC es similar a las cepas AIEC en términos de capacidad de crecimiento. Las predicciones de crecimiento sugieren que las cuatro cepas (CG1MAC, K-12, LF82 y NC101) tienen capacidades metabólicas distintas, ya que su capacidad de crecimiento predicha difiere para 35 sustratos (Figura 5A). El fenotipo de crecimiento predicho mostrado por CG1MAC es similar al de LF82 y NC101, ya que comparten la capacidad de utilizar un subconjunto de seis sustratos (marcados en naranja en la Figura 5A), pero no K-12. Entre los seis sustratos identificados, algunos se encuentran abundantemente en el intestino, incluida la celobiosa, un derivado de una celulosa de fibra dietética insoluble (Cummings, 1984; Cocinero et al., 2009), así como monosacáridos derivados de la mucosa intestinal: N-acetil-D-galactosamina (GalNAc) y N-acetil-D-galactosamina 1-fosfato (GalNAc 1P) (Ravcheev y Thiele, 2017). La capacidad de utilizar desoxirribosa, por otro lado, sugiere patogenicidad de CG1MAC y NC101. Un estudio previo demostró que la capacidad de metabolizar la desoxirribosa se asocia con el potencial patogénico de las cepas intestinales y extraintestinales de E. coli, ya que esta capacidad aumenta su competitividad (Bernier-Fébreau et al., 2004). La disponibilidad de desoxirribosa también promueve la colonización del huésped del intestino por cepas patógenas de E. coli (Martinez-Jéhanne et al., 2009). Los dos sustratos restantes, el 3-fosfo-D-glicerato (3PG) y el 2-fosfo-D-glicerato (2PG), son intermediarios importantes en la glucólisis (Neidhardt y Curtiss, 1999) y precursores de la biosíntesis de aminoácidos (Kaleta et al., 2013). La capacidad de absorber directamente estos sustratos permite potencialmente que NC101 y CG1MAC generen energía de manera más eficiente, por lo que es probable que superen a otros microbios. También hemos identificado reacciones que permiten el crecimiento en los seis sustratos anteriores, que faltan en K-12 (etiquetados en rojo en la Figura 5B). Observamos que K-12 carece de transportadores para los seis sustratos, así como de algunas enzimas aguas abajo. También realizamos experimentos experimentales de crecimiento para la validación del modelo (ver Material Suplementario y Tabla S4).
Figura 5
Figura 5. Resultados de simulación de cuatro GEMs. (A) Las capacidades de crecimiento de varias fuentes de nutrientes se pueden utilizar para diferenciar entre cepas. (B) Las vías clave involucradas en la capacidad de catabolir los seis sustratos destacados. Las enzimas en rojo están ausentes en E. coli K-12.
3. Discusión
En este estudio realizamos un análisis metagenómico a nivel de cepa de E. coli en una serie temporal de muestras de heces de un paciente con EC. Los hallazgos clave son los siguientes: (1) La comunidad de E. coli fue altamente dinámica en este paciente, con diferente abundancia relativa y cepas dominantes en diferentes momentos. (2) Pudimos extraer ensamblajes de novo a nivel de cepa de siete cepas dominantes a partir de datos metagenómicos, y mostramos una gran variación en el contenido genómico entre las cepas utilizando un análisis pangenómico. (3) El análisis genómico comparativo y la reconstrucción de la red metabólica sugieren que ST1 (aislada durante el pico de inflamación) se asemeja a las cepas de referencia conocidas de AIEC NC101 y LF82, mientras que otras cepas recolectadas durante estados estables mostraron características diversas. (4) Para evaluar la precisión de los ensamblajes de novo a partir de datos metagenómicos, aislamos ST1 (denominado CG1MAC) de la muestra de heces, lo secuenciamos y lo caracterizamos experimentalmente. (5) A continuación, construimos un modelo metabólico completo a escala genómica de CG1MAC y evaluamos su capacidad de crecimiento.
Los datos detallados de series temporales no solo mostraron la disbiosis intestinal de este paciente, sino que también revelaron la dinámica de su microbioma intestinal a nivel de cepa. Aunque estudios recientes ya han mostrado fluctuaciones drásticas tanto en la composición como en la función del microbioma intestinal de los pacientes con EII (Halfvarson et al., 2017; Schirmer et al., 2018), y lo vincularon con el desarrollo de enfermedades (Sharpton et al., 2017), solo se centraron en las evaluaciones a nivel de especie. En este estudio, sin embargo, presentamos la dinámica a nivel de cepa de la comunidad de E. coli: no solo la abundancia relativa de E. coli varió con el tiempo, sino que también identificamos siete cepas que dominaron la comunidad de E. coli en diferentes puntos de tiempo, que luego se demostró que tenían diversos contenidos genéticos y orígenes filogenéticos mediante ensamblajes de novo.
El análisis a nivel de cepa de las cepas dominantes de E. coli y su correlación con los metadatos nos llevó a plantear la hipótesis de que solo ciertas cepas de E. coli con características específicas contribuyen a la progresión de la EII. El análisis genómico comparativo y las reconstrucciones de redes metabólicas sugieren similitud tanto en los factores de virulencia como en las funciones metabólicas entre ST1 (recolectado durante el pico de inflamación) y los aislados de EII patógena conocidos NC101 y LF82. La evidencia de la literatura sugiere que el patotipo AIEC, al que pertenecen tanto LF82 como NC101, está implicado en la EII. Sin embargo, aislamos y caracterizamos experimentalmente ST1 (más tarde llamado CG1MAC), y encontramos que no muestra fenotipos AIEC. Curiosamente, estudios anteriores centrados en aislamientos clínicos también han aislado cepas no AIEC de pacientes con EII, así como cepas AIEC de controles sanos (O’Brien et al., 2016). Estos resultados sugieren que las cepas capaces de provocar una respuesta inflamatoria en pacientes con EII pueden compartir ciertas características, pero no necesariamente pertenecer al patotipo AIEC. Aunque este resultado debe verificarse más a fondo, tanto experimentalmente como en una cohorte más grande, ilustra la importancia de las evaluaciones del microbioma intestinal a nivel de cepa. Otro aspecto que debe tenerse en cuenta en futuros estudios es la asociación entre las características específicas de la cepa y los subtipos de EII (EC ileal, EC colónica y colitis ulcerosa), ya que la investigación ha demostrado que los tres subtipos están determinados genéticamente y pueden ser desencadenados por diferentes factores externos (Cleynen et al., 2016).
Además, con la secuencia de CG1MAC, confirmamos la validez de los ensamblajes de novo y caracterizamos la capacidad de crecimiento de CG1MAC con un GEM preciso. Los ensamblajes de novo a nivel de cepa no se han adoptado ampliamente en los estudios del microbioma, pero ilustramos el potencial y la viabilidad de dicho análisis, ya que el ensamblaje ST1 captura con precisión el 95,5% del contenido real del genoma. Por otro lado, otra poderosa herramienta, los GEM, nos permitió predecir que: CG1MAC, junto con NC101 y LF82, son capaces de utilizar sustratos que son abundantes en el intestino humano (incluyendo celobiosa y glicanos mucosos), o sustratos que potencialmente les permiten superar a otras cepas como la desoxirribosa (Bernier-Fébreau et al., 2004; Martínez-Jéhanne et al., 2009).
Además, la medicación también desempeña un papel importante en la composición del microbioma intestinal. Se ha demostrado que los antibióticos, como la ciprofloxacina y el metronidazol, reducen la diversidad bacteriana y disminuyen la abundancia de enterobacterias (Langdon et al., 2016), mientras que los corticosteroides como la prednisona y el uceris, pueden contribuir a un cambio sustancial en la microbiota intestinal (Huang et al., 2015). Además, este paciente también ha tomado mesalamina (Lialda) que ha demostrado disminuir la abundancia de Escherichia y Shigella (Morgan et al., 2012). Observamos en este paciente que después de tomar Ciprofloxacino, Metronidazol y Prednisona en febrero de 2012, el nivel de PCR disminuyó drásticamente, mientras que la diversidad alfa también disminuyó (Figura S3). Después de tomar Uceris y Liada en 2013 de junio a noviembre, no se observó más sangrado ni brote. Sin embargo, se necesitan más datos y experimentos para obtener una comprensión completa del impacto de los medicamentos en la estructura del microbioma y la progresión de la enfermedad.
También reconocemos algunas limitaciones de este enfoque que deben abordarse en el futuro: (1) La precisión del ensamblaje de novo a nivel de cepa a partir de datos metagenómicos debe evaluarse cuidadosamente. Nuestro estudio mostró que dicho ensamblaje no captura la secuencia del genoma con una precisión del 100%, y dicho análisis solo es posible para muestras con una alta cobertura de lectura de E. coli. Sin embargo, con el rápido desarrollo de las herramientas de análisis de metagenómica, se espera que la calidad y la viabilidad del ensamblaje de novo a nivel de cepa mejoren en el futuro. (2) En este estudio solo examinamos los datos de metagenómica, no el nivel de expresión génica. Al incluir la metatranscriptómica en el futuro, uno debería ser capaz de describir los estados funcionales de los microbios con mayor precisión. (3) Este flujo de trabajo solo nos permite examinar las cepas dominantes en cada punto de tiempo, mientras que las cepas de E. coli de menor abundancia no se tienen en cuenta. Por lo tanto, no se caracteriza la variación genética en la comunidad de E. coli en cada momento. (4) Es necesario tener en cuenta otros factores que contribuyen a la EII. La asociación entre las características de las cepas de E. coli y otros elementos, como la genómica del huésped, la dieta y sus vecinos microbianos, probablemente agregará información valiosa para futuros análisis. En general, creemos que realizar un análisis de este tipo en una gran cohorte de pacientes con EII enriquecerá en gran medida nuestro conocimiento de la EII y el microbioma intestinal.
4. Conclusiones
En este estudio, observamos que la cepa dominante de E. coli en este paciente varió con el tiempo. En particular, las cepas dominantes aisladas durante el pico de inflamación son más similares a las cepas patógenas conocidas implicadas en la EII, mientras que otras cepas recolectadas durante estados más estables tienen propiedades diversas. En general, este estudio piloto ilustra que un análisis a nivel de cepa de E. coli a partir de una serie temporal de muestras de heces puede ser muy productivo. El enfoque que utilizamos en este estudio no solo captura la estructura y la dinámica de toda la comunidad del microbioma, sino que también permite una evaluación detallada de E. coli a nivel de cepa. Debido a la disminución del coste de la secuenciación y a la menor cantidad de procedimientos experimentales involucrados, este enfoque también debería permitir análisis rápidos y a gran escala en el futuro.
5. Materiales y métodos
5.1. Generación de datos metagenómicos
El ADN se extrajo de muestras fecales primarias utilizando el kit de extracción MoBio PowerMag (Qiagen Inc). Las bibliotecas metagenómicas de escopeta se prepararon y secuenciaron en las instalaciones centrales de secuenciación del Instituto de Medicina Genómica de la UCSD. Brevemente, se construyeron bibliotecas a partir de cada muestra utilizando 200 ng de ADN extraído, cortadas a un tamaño de fragmento objetivo de aproximadamente 400 pb utilizando un sonicador Covaris E220 y entrada en el kit de preparación de bibliotecas basado en PCR TruSeq Nano (Illumina Inc), con muestras indexadas individualmente utilizando adaptadores duales con código de barras de 8 pb. A continuación, las bibliotecas amplificadas se agruparon y secuenciaron en un instrumento HiSeq4000. Las lecturas secuenciadas se recortaron de las secuencias adaptadoras y se filtraron por calidad utilizando Skewer (Jiang et al., 2014) (parámetro de recorte de calidad final de Phred 15 y un ajuste de longitud mínima de 100 pb después del recorte) y cutadapt (Martin, 2011) v1.15 (parámetros -m 36 -q 20 -a ATCGGAAGAGCACACGTCTGAACTCCAGTCAC, -A ATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTGT). A continuación, se filtraron las secuencias recortadas de las lecturas derivadas de humanos utilizando Bowtie2 (Langmead y Salzberg, 2012) en la configuración “muy sensible”, conservando solo los pares de lectura para los que ninguno de los dos pares se asignó a la referencia humana.
5.2. Análisis de datos metagenómicos
Los perfiles taxonómicos de los datos metagenómicos se evaluaron utilizando MetaPhlan2 (Truong et al., 2017) con parámetros predeterminados. Extrajimos la abundancia relativa de E. coli del resultado y la comparamos entre muestras. Además del análisis de MetaPhlan2, también realizamos análisis adicionales para confirmar la presencia de bacteriófagos en una muestra utilizando Bowtie2 mediante el mapeo de las lecturas de secuenciación a las secuencias del genoma de los dos fagos (Langmead2012-zv) (ver Material suplementario).
La diversidad alfa y beta de los datos metagenómicos se calculó con el paquete de python skbio (SCI, 2018). Utilizamos los perfiles taxonómicos previamente calculados a nivel de especie como tablas de entrada de OTU para el cálculo de la diversidad. Se calculó la diversidad alfa utilizando la métrica “observed_otus” y la diversidad beta utilizando la métrica “braycurtis”. A continuación, realizamos un análisis de coordenadas principales y trazamos los PC1, 2 y 3 utilizando el mismo paquete de python skbio.
5.3. Caracterización de las cepas dominantes de E. coli utilizando frecuencias de variantes de un solo nucleótido (SNV)
En primer lugar, se utilizó la tubería MIDAS (base de datos v 1.2) (Nayfach et al., 2016) con valores predeterminados para llamar a los SNV de todo el genoma para todas las especies abundantes dentro de las muestras individuales. La información de frecuencias de SNVs para E. coli 58110 (genoma representativo de especies de E. coli en la base de datos MIDAS) se fusionó en todas las muestras. La Figura S4 ilustra las frecuencias alélicas menores en sitios genómicos particulares en todas las muestras (posiciones elegidas por MIDAS, columnas reordenadas con respecto a su agrupamiento jerárquico). El mapa de calor sugiere que la población de E. coli dentro de la mayoría de las muestras metagenómicas estaba dominada por una sola cepa (solo se observan valores cercanos a 0 o 1 en las filas respectivas) que cambiaron con el tiempo.
A continuación, realizamos un análisis computacional refinado de las frecuencias de SNV para confirmar esta hipótesis e identificar muestras con la misma cepa dominante de E. coli. Para cada muestra metagenómica, se utilizó la tubería MIDAS con el parámetro “–species_id Escherichia_coli_58110” para calcular la cobertura por base y las frecuencias de SNV para la referencia de E. coli. Para evitar varios artefactos, descartamos los sitios con cobertura aberrante de la siguiente manera: posiciones con cobertura 0, posiciones con cobertura inferior al doble de la mediana en los sitios restantes y posiciones con cobertura que cae dentro del 10% bajo/alto de los valores de cobertura en los sitios restantes.
Para probar si la población de E. coli dentro de la muestra está dominada por una sola cepa, analizamos las frecuencias alélicas variantes en las posiciones restantes. En concreto, se consideró que la población estaba dominada si menos del 0,05% de las posiciones tenían una frecuencia de variantes menores superior al 10%. Todas las muestras, excepto dos (TP23 y TP27) cumplieron esta condición.
Además, intentamos dividir las 19 muestras restantes en grupos dominados por la misma cepa. Definimos la similitud entre el par de muestras como una fracción de las posiciones en las que coincidían las variantes principales (solo se consideraron los sitios retenidos en el análisis de ambas muestras). Se utilizó el agrupamiento de enlace simple con un umbral del 99,9% para obtener 7 grupos de muestras, cada uno correspondiente a una cepa particular de E. coli. Se ha elegido una sola muestra dentro de cada grupo para intentar la reconstrucción del genoma de la cepa mediante ensamblaje de novo (ver sección 5.4).
5.4. Ensamblaje de cepas dominantes de E. coli a partir de datos metagenómicos
Se ha utilizado el ensamblador metaSPAdes v3.11.1 con parámetros predeterminados para realizar el ensamblaje de novo de 7 muestras metagenómicas individuales (28/12/2011; 3/4/2012; 7/8/2012; 14/7/2013; 23/3/2014; 25/8/2014; 28/9/2014).
Los andamios resultantes y sus profundidades de cobertura (cobertura promedio de 56 meros reportada por metaSPAdes) se proporcionaron como entrada para MaxBin2 (Wu et al., 2016). Cada muestra contenía un contenedor anotado como E. coli con una completitud estimada superior al 97% (según lo informado por CheckM Parks et al., 2015), que se utilizó como borrador de ensamblaje para el análisis posterior. También hemos considerado incluir contenedores más pequeños anotados como E. coli por MaxBin2, pero ha resultado en un fuerte aumento del nivel de contaminación (según lo informado por CheckM). Las puntuaciones de contaminación e integridad de estos conjuntos se informan en la Tabla S5.
5.5. Análisis filogenético y construcción pangenómica de los siete ensamblajes
En primer lugar, anotamos los ensamblajes utilizando Prokka (Seemann, 2014) con parámetros predeterminados. Los archivos de salida de Prokka se utilizaron para realizar análisis filogenéticos y reconstrucción del pangenoma. Para realizar el análisis filogenético utilizando PhyloPhlan (Segata et al., 2013), utilizamos los archivos de proteínas FASTA terminados en “.faa” de la salida de Prokka, y construimos los árboles filogenéticos con otras 110 cepas de E. coli con filogrupos conocidos para inferir el filogrupo de cada ensamblaje. Para construir el pangenoma de los siete ensamblajes, utilizamos Roary (Page et al., 2015) que toma archivos de entrada terminados en “.gff”, que contienen la anotación maestra en formato GFF3 producida por Prokka. Establecemos el parámetro “identidad porcentual mínima para blastp” en 80.
5.6. Análisis del factor de virulencia
Mapeamos los ensamblajes del genoma de las cepas dominantes contra dos conjuntos de referencias de factores de virulencia. El primer conjunto son los factores de virulencia seleccionados de la base de datos VFDB (Chen et al., 2005). El segundo conjunto son 57 genes identificados en la literatura que están asociados con cepas de AIEC, que están implicadas en la EII. Estos genes se identifican y recolectan principalmente de acuerdo con el artículo de revisión de Palmela et al. (2017). Tenga en cuenta que los 57 genes contienen variantes de genes que realizan las mismas funciones. Utilizamos BLAST (Boratyn et al., 2012) para mapear los ensamblajes a las referencias y consideramos que los genes están presentes cuando la similitud de secuencia es superior al 80%.
5.7. Reconstrucción de la red metabólica y análisis de la matriz pan-reactoma
El borrador de las reconstrucciones metabólicas de las cepas de E. coli se crea en base a un estudio previo de múltiples cepas de E. coli (Monk et al., 2013). Primero creamos un modelo de E. coli que combina todos los genes, reacciones y metabolitos en los 55 modelos de E. coli reconstruidos por Monk et al. (2013). Para incorporar la actualización más reciente en la reconstrucción de E. coli, también agregamos el contenido del último modelo K-12 iML1515 (Monk et al., 2017) al modelo de bandeja. Dado que todas las cepas de E. coli incluidas abarcan varios patotipos y orígenes filogenéticos, el modelo de pan creado se considera una representación completa de las funciones metabólicas en las cepas de E. coli, así como un buen punto de partida para la reconstrucción de la red metabólica. A continuación, mapeamos las secuencias de cepas de interés para todos los genes en el modelo pan, utilizando BLAST (McGinnis y Madden, 2004), y establecimos un umbral del 80% tanto para la similitud génica como para la longitud de alineación, con el fin de que un gen se considerara presente en las cepas. Los genes faltantes y sus reacciones y metabolitos correlacionados en cada cepa se eliminan del modelo pan para crear reconstrucciones de redes metabólicas específicas de la cepa. La red metabólica se reconstruyó utilizando el paquete de python COBRApy (Ebrahim et al., 2013).
Para comparar las redes metabólicas de las 7 cepas dominantes y las 3 cepas de referencia, creamos una matriz binaria de tamaño 10 por 3.077 que registra la presencia y ausencia de cada reacción en las 10 cepas. Para determinar la similitud en las funciones metabólicas en 10 cepas, se realizó un análisis de ACM utilizando el paquete mca de python (mca, 2018) con corrección de Benzecri, con el parámetro de TOL establecido en 1e-9. Para extraer las reacciones importantes en el factor 1 y el factor 2, identificamos las 50 reacciones principales que tienen la mayor contribución a estos dos factores (Tabla S3).
5.8. Aislamiento de cepas bacterianas: CG1MAC y 3_2_53FAA
Con el fin de aislar CG1MAC de la muestra de heces, diluimos la muestra en solución salina y se sembraron diluciones en agar McConkey para seleccionar los aislamientos de E. coli. Se seleccionaron todos los aislados obtenidos y se extrajo el ADNg utilizando un mini kit de heces Qiagen. Para verificar los aislamientos que se identificaron con el genotipo predicho de la cepa diana, se utilizaron cuatro genes, fyuA, vasD, xerD, gsp, a los que se diseñaron cebadores de PCR basados en datos de secuencia del ensamblaje de novo de ST1. El análisis comparativo mostró que estos genes estaban presentes en el conjunto de datos metagenómicos obtenidos de la muestra de heces de origen y son más prevalentes en las cepas de E. coli asociadas a la EII. Se obtuvieron 40 cepas y se tamizaron por PCR de esta manera, y se encontró que todas se identificaron positivamente con el ensamblaje ST1. De los clones, uno fue seleccionado para un análisis más detallado, y se denominó CG1MAC.
La cepa 3_2_53FAA se aisló a partir de una muestra de biopsia inflamada del colon descendente de un paciente masculino de 52 años con EC del lado izquierdo en una clínica de Calgary, Canadá, en 2007. El paciente tuvo un diagnóstico inicial de colitis ulcerosa que posteriormente se cambió a colitis de Crohn (las biopsias ileales fueron normales). La cepa 3_2_53FAA se colocó en la colección de genomas de referencia del Proyecto Microbioma Humano como HM-38 y, como tal, fue secuenciada por el Instituto Broad (número de acceso al ensamblaje GenBank GCA_000157115.2).
Tanto CG1MAC como 3_2_53FAA fueron serotipificados por el Laboratorio Nacional de Microbiología (Agencia de Salud Pública de Canadá) en Guelph, Ontario.
5.9. Secuencia del genoma bacteriano
Secuenciamos el genoma de la cepa aislada de E. coli CG1MAC. En primer lugar, aislamos y purificamos el ADNg de las células peletizadas utilizando el kit de tejido Macherey-Nagel NucleoSpin (número de catálogo 740952.50) siguiendo el protocolo del fabricante, incluido el tratamiento con ARNasa. En segundo lugar, preparamos una biblioteca de ADN genómico utilizando un kit de preparación de bibliotecas KAPA HyperPlus (número de catálogo KK8514) que incorporaba índices duales durante la etapa de amplificación de la PCR y comprobaba la calidad con TapeStation. Finalmente, agrupamos la biblioteca y la secuenciamos utilizando el instrumento Illumina HIseq 4000 con configuraciones de extremo emparejado y lecturas 100/100.
Utilizamos SPAdes (Bankevich et al., 2012) para ensamblar las lecturas de alta calidad con parámetros predeterminados. El genoma ensamblado ha sido presentado al NCBI con el número de acceso QLAC00000000.
5.10. Confirmación de la identidad aislada de CG1MAC con análisis de SNV
Utilizamos el análisis de frecuencias de variantes de un solo nucleótido (SNV) de todo el genoma para verificar que: (1) la población de E. coli en la muestra metagenómica TP1 está dominada por una sola subpoblación; (2) La subpoblación dominante está representada por la cepa aislada CG1MAC.
Las lecturas de aislados de TP1 y CG1MAC se procesaron mediante la tubería MIDAS (Nayfach et al., 2016) con el parámetro –species_id Escherichia_coli_58110 para calcular la cobertura y las frecuencias de SNV para las lecturas de secuenciación metagenómica y aislada contra la referencia de E. coli incluida en su base de datos.
En primer lugar, demostramos que la población de E. coli en TP1 probablemente esté dominada por una sola cepa. Para evitar varios artefactos, ignoramos las posiciones con cobertura que se encuentran dentro del 10% bajo/alto de los valores de cobertura en todas las posiciones cubiertas de la referencia. De los 2,85 millones de sitios restantes, solo 181 tenían una frecuencia de alelos mayores (MAF) que no superaba el 90% (en comparación, la muestra de CG1MAC tenía 96 de esas posiciones), lo que sugiere que una sola cepa representó la mayor parte de la población de E. coli. A continuación, comparamos los genotipos predichos del aislado CG1MAC y la cepa dominante de E. coli en TP1. Solo se consideraron los sitios con MAF ≥ 90% y la cobertura dentro de los percentiles 10 y 90 en ambas muestras. Si bien cubren el 59% del genoma de referencia (total 2,48 Mb), no se observaron diferencias entre los alelos principales de las dos muestras, lo que indica de manera confiable que el aislado CG1MAC se origina en la subpoblación dominante.
5.11. Curación del modelo CG1MAC
En primer lugar, se creó el borrador de reconstrucción metabólica de CG1MAC siguiendo el procedimiento descrito en la sección 5.6. A continuación, realizamos una selección adicional para mejorar la precisión del borrador del modelo. Anotamos el genoma de CG1MAC con RAST (Aziz et al., 2008) e identificamos genes metabólicos utilizando números de Comisión Enzimática (EC). A continuación, identificamos 413 genes metabólicos no incluidos en el modelo pan, y analizamos las reacciones asociadas a ellos en la base de datos Uniprot (The UniProt Consortium, 2017) en relación con su puntuación de anotación y la evidencia experimental. Entre todas las reacciones identificadas, solo agregamos seis al modelo en función de los siguientes criterios de filtrado: (1) Tener un número EC completo con cuatro números; (2) No está involucrado en la modificación del ADN/ARN, como sugiere el protocolo de reconstrucción GEM establecido (Thiele y Palsson, 2010); (3) se ha demostrado experimentalmente que está presente en E. coli; (4) tener una reacción definida con especificidad; (5) No duplique con las reacciones existentes. La mayoría de las reacciones identificadas ya están presentes en el modelo, ya que sus genes codificantes son variantes de genes existentes en el modelo. A continuación, añadimos las nuevas reacciones al modelo CG1MAC y a los 3 modelos de referencia cuando correspondía, para garantizar que la simulación de crecimiento realizada en estos cuatro modelos sea precisa. Finalmente, se realizó el paso de curación manual para el modelo CG1MAC siguiendo el protocolo establecido (Thiele y Palsson, 2010). Debido a que los 55 modelos existentes en los que se basó la reconstrucción ya estaban seleccionados manualmente, nos centramos en seleccionar las reacciones recién agregadas. Eliminamos las reacciones y los metabolitos en el compartimento equivocado, añadimos el subsistema de las nuevas reacciones, nos aseguramos de que las nuevas reacciones estuvieran equilibradas entre masa y carga y comprobamos la reacción gen-proteína (gpr) de las reacciones recién añadidas.
5.12. Ensayos de adhesión e invasión en células Caco-2 y THP-1
Para determinar la invasión bacteriana en células epiteliales y la supervivencia en macrófagos Caco-2, las células se mantuvieron en DMEM + 10% FBS (Invitrogen). Las células THP-1 se mantuvieron en RPMI + 10% FBS (Invitrogen) en atmósfera humidificada con CO2 al 5% a 37°C. La diferenciación de las células THP-1 se logró mediante el tratamiento con 5 ng/ml de PMA (Sigma-Aldrich) durante 2 días. Se permitió que las células se recuperaran en medios normales durante 1 día antes de realizar el ensayo.
Los ensayos de adhesión, invasión y supervivencia se realizaron como se describe en Negroni et al. (2012). Brevemente, se realizaron análisis de invasión celular en células de Caco-2 cultivadas en DMEM sin antibióticos, y mantenidas en 5% CO2 y 37°C. Las monocapas celulares se infectaron con cepas de E. coli a una multiplicidad de infección (MOI) de 100, durante 2 h a 37 °C. Después del período de infección, las células se lavaron con 3 x PBS y se colocaron en medio fresco suplementado con gentamicina (50 μg/ml), se incubaron durante 1 h a 37 °C y se lisaron con Triton-X-100PBS al 0,1%. Las diluciones seriadas de lisado se sembraron en agar LB (Invitrogen) y se incubaron a 37 °C durante la noche. También se llevó a cabo un análisis de adhesión celular en células Caco-2 utilizando condiciones de infección similares a las descritas para los ensayos de invasión, pero omitiendo el tratamiento con gentamicina. Las células diferenciadas de THP-1 se infectaron con cepas de E. coli (MOI = 100) durante 2 h a 37 °C. A continuación, las células se lavaron en PBS y se colocaron en un medio fresco suplementado con gentamicina (50 μg/ml). Se determinó el contenido bacteriano intracelular a 1 y 24 h post infección a 37°C y se determinó la relación entre el contenido bacteriano en cada período.
5.13. Simulaciones de crecimiento in silico
La simulación de crecimiento para CG1MAC, K-12, LF82 y NC101 se realizó utilizando COBRApy. Simulamos el crecimiento en medios mínimos M9, con el límite inferior de las reacciones de intercambio para el siguiente sustrato establecido en -1000: Ca2+, Cl−, CO2, Co2+, Cu2+, Fe2+, Fe3+, H+, H2O, K+, Mg2+, Mn2+, MoO42MoO42, Na+, Ni2+, SeO42−SeO42−, SeO32+SeO32+ y Zn2+. Además, las fuentes predeterminadas de carbono, nitrógeno, azufre y fosfato son glucosa, NH4−NH4−, SO42SO42, HPO42HPO42. Estas reacciones tienen límites inferiores establecidos en -1000. Otro sustrato esencial es la cob(I)alamina, para la cual la reacción de intercambio tiene un límite inferior de -0,01. Evaluamos si el sustrato de carbono, nitrógeno, azufre o fosfato soportaba el crecimiento. Para ello, establecemos el límite inferior de la reacción de intercambio del sustrato predeterminado en 0 y agregamos el único sustrato estableciendo el límite inferior de la reacción de intercambio en −10. Además, hemos simulado el crecimiento en condiciones aeróbicas estableciendo el límite inferior de absorción de oxígeno en -10.
5.14. Experimentos de crecimiento
Las cepas de E. coli K12 y CG1MAC se cultivaron en medios M9 modificados con la fuente principal de carbono, nitrógeno o azufre reemplazada. Para las pruebas que implicaban la sustitución de la fuente de carbono, se omitió la glucosa del medio M9 y en su lugar se añadieron 0,022 moles/L de la nueva fuente de carbono. Para las pruebas de reemplazo de fuente de nitrógeno, se reemplazó NH4Cl por 0.019 moles/L de la nueva fuente de nitrógeno y para las pruebas de reemplazo de fuente de azufre, HPO4 • 7 H2O se reemplazó por 0.001 moles/L de la nueva fuente de azufre. Para los medios utilizados en las pruebas de reemplazo de nitrógeno y azufre, la concentración de glucosa se incrementó a 0,004 g/mL.
Las colonias individuales recién cultivadas de cada cepa se seleccionaron después de la incubación durante la noche en placas de agar sangre y se diluyeron individualmente en 5 mL de medio M9 basal (sin fuente de carbono, nitrógeno o azufre) y se utilizaron 100 μL de cada cepa diluida para inocular 5 mL de medio M9 modificado que contenía la fuente de carbono, nitrógeno o azufre de prueba. Los tubos inoculados se incubaron durante 24 h a 37°C con agitación orbital a 200 rpm para preexponer las células a cada metabolito. Después de la incubación, las células se peletizaron a 5.000 rpm durante 10 min y se resuspendieron en tampón PBS de 200 μL, tras lo cual se registró la densidad óptica a 555 nm. Este valor se utilizó para normalizar la cantidad de cada cultivo que se añadió a 5 ml del medio de ensayo apropiado en un tubo de ensayo de vidrio. Los tubos de ensayo de muestra se incubaron durante 48 h a 37 °C con agitación orbital a 200 rpm, y se utilizaron muestras de 100 μL de estos tubos para medir la densidad óptica a 555 nm, que se registró cada 24 h utilizando un lector de placas Wallac Victor 3.
Los metabolitos utilizados fueron: glucosa (Fisher Scientific); melibiosa, fenilacetaldehído, ácido trans-3-hidroxicinámico, ácido oxaloacético, clorhidrato de dopamina, 2-desoxi-D-ribosa, citrato de hierro (III), taurina, treonina y tiosulfato de sodio (de Sigma Aldrich); celobiosa y D-arabinosa (de Fluka); Propionato de 3-(3-hidroxi-fenil), D-arabinosa, cloruro de colina, D-(+)-galactosa, ácido 3-hidroxifenilacético, tiramina y acetato de metil-4-hidroxifenilacetato (de Alfa Aesar) y sacarosa (de Bioshop).
5.15. Declaración de ética
El paciente al que se le recogieron las muestras de heces tiene su consentimiento bajo dos protocolos: HRPP #141853 American Gut Project y HRPP #150275 Evaluación del microbioma humano. Ambos protocolos fueron aprobados por el Programa de Protección de la Investigación Humana (HRPP, por sus siglas en inglés) de la Universidad de California en San Diego. El consentimiento informado por escrito sobre la difusión del resultado y la publicación científica también se incluyen en los protocolos aprobados, y se obtuvo del paciente.
El paciente en el que se aisló la cepa 3_2_53FAA fue reclutado y consentido a través del Banco de Tejidos de Inflamación Intestinal de la Universidad de Calgary y este estudio fue aprobado a través de la Junta de Ética de Investigación en Salud Conjunta de la Universidad de Calgary (Números de Proyecto; REB14-2429 y REB14-2430).
Declaración de disponibilidad de datos
La secuencia metagenómica de las 27 muestras se ha enviado a EBI en estudio PRJEB24161. Tenga en cuenta que estas muestras son solo un subconjunto de los datos metagenómicos de este estudio. La secuencia del genoma de CG1MAC se puede encontrar en el NCBI con el número de acceso QLAC00000000.